
Christopher Burge, MIT
Eric Wang
Daniel Treacy
Overall study coordination, preparation of deep sequencing libraries, data analysis, website development
Deep Sequencing
Data Repository
A collaboration to comprehensively survey the myotonic dystrophy transcriptome

Myotonic dystrophy (dystrophia myotonica, "DM") has been described as "one of the most variable of all human disorders" (Peter Harper). It is a microsatellite repeat expansion disorder, and similar to other repeat expansion diseases, exhibits genetic anticipation. This phenomenon commonly leads to earlier age of onset and more severe symptoms for children who inherit the expansion from affected individuals. Both germ line and somatic instability likely lead to variability in DM symptoms, which include muscle weakness and wasting, myotonia, cardiac arrhythmia, profound fatigue, gastrointestinal distress, and cognitive deficits, among others.
While understanding the molecular basis for such a variable disease is challenging, the scientific community has successfully identified several disease mechanisms that clearly play a role in DM. Gene expression and RNA processing are perturbed in DM, and both processes can be better understood through the study of transcriptomes from human samples and animal models. With the help and support of generous collaborators, and most importantly, patients who have donated precious biological material, we are able to provide here transcriptome data from DM biopsies, autopsies, and animal models. We hope that this repository and its associated tools will be a valuable resource for the scientific community, so as to accelerate the path towards basic understanding and treatment of DM and related diseases.
This project was supported by NIH ARRA grant 5RC2HG005624-02, awarded to principal investigators Christopher Burge, David Housman, and Thomas Cooper. It would not have been possible without the generous contributions of numerous collaborators.
Overall study coordination, preparation of deep sequencing libraries, data analysis, website development

Study coordination, RNA isolation from tissue

Study coordination, tissue from mouse models of DM and human autopsies

Human biopsy RNA

Human biopsy tissue

Human autopsy tissue

Data analysis, website development
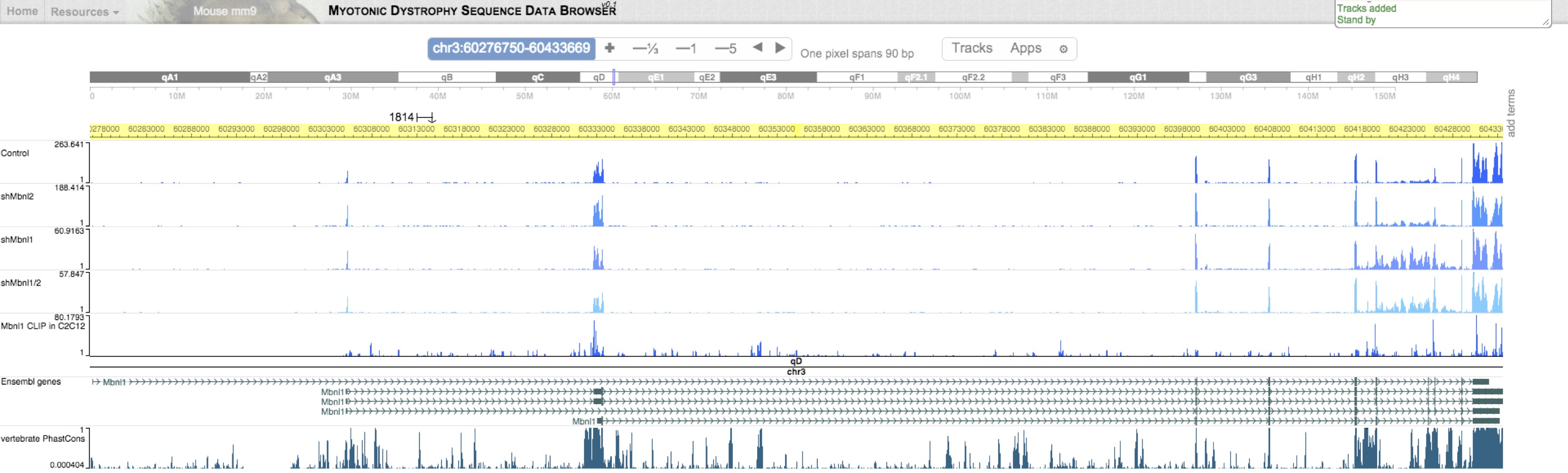
This genome browser is powered by the WashU Epigenome Browser. Raw read density across the genome can be loaded as "Public tracks". To view human data, select "hg19". To view mouse data, select "mm9". Additional transcriptomes from related studies are also included as "Public tracks". While you can load your own "custom tracks", contact us to request specific sets of tracks loadable as "data hubs".
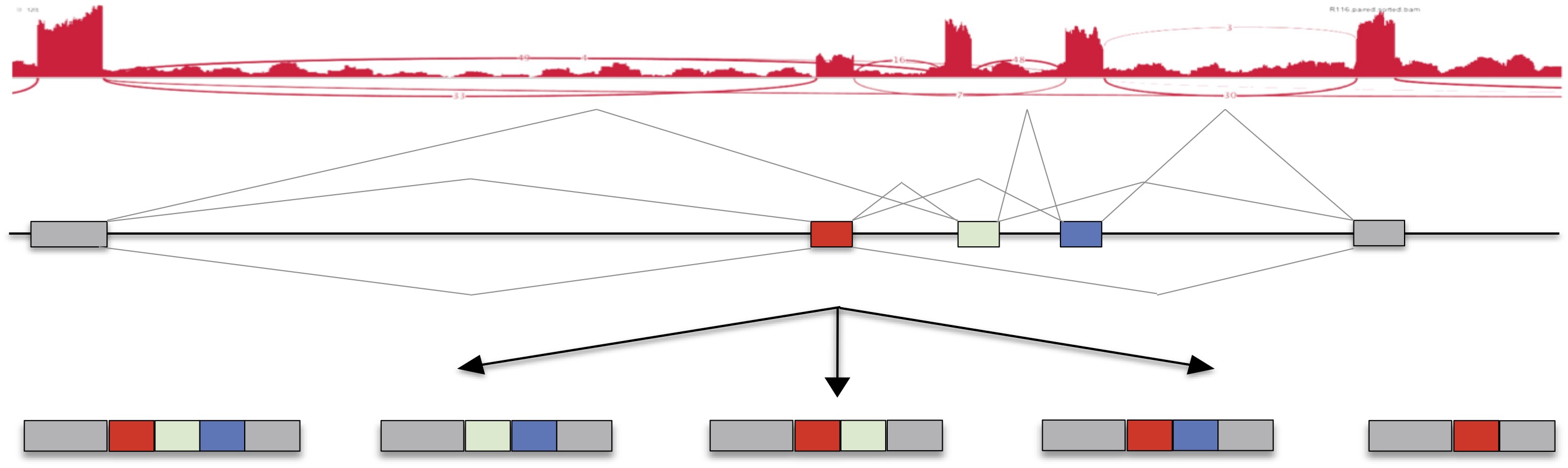
Gene expression values, and isoform expression as estimated by MISO, can be viewed here. Search for specific genes, and generate bar plots and sashimi plots.

Alignment (BAM), bedgraph, MISO output, and other data files may be downloaded here.
Please don't hesitate to submit your questions and comments. Your feedback is appreciated.